publications
publications by categories in reversed chronological order. generated by jekyll-scholar.
2026
-
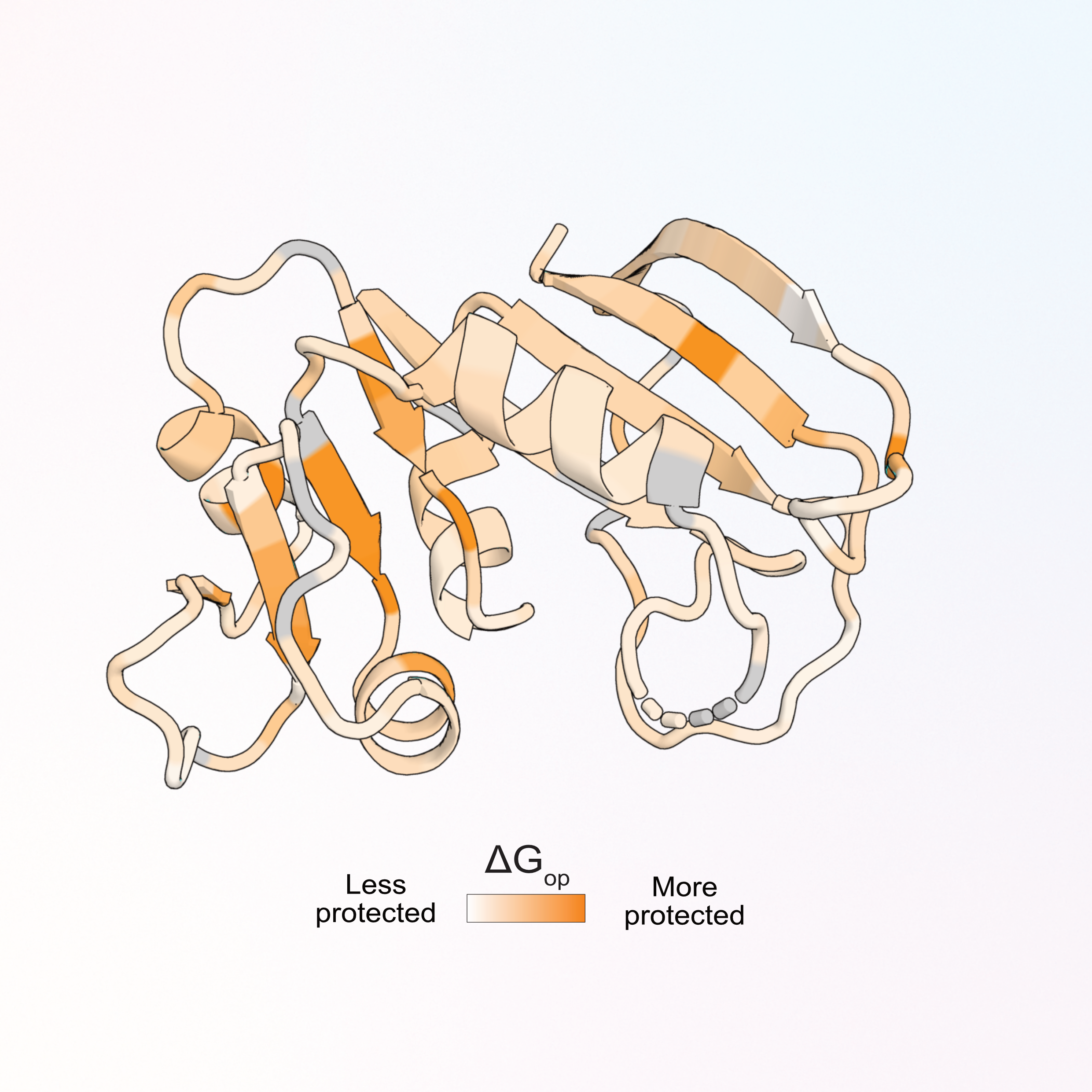 Site-resolved energetic information from HX-MS experimentsChenlin Lu*, Malcolm L. Wells*, Andrew Reckers, and 2 more authorsNature Chemical Biology, 2026
Site-resolved energetic information from HX-MS experimentsChenlin Lu*, Malcolm L. Wells*, Andrew Reckers, and 2 more authorsNature Chemical Biology, 2026While bioinformatics reveals patterns in protein sequences and structural biology methods elucidate atomic details of protein structures, it is difficult to attain equally high-resolution energetic information about protein conformational ensembles. We present PIGEON-FEATHER, a method for calculating free energies of opening (ΔGop) at single- or near-single-amino acid resolution for protein ensembles of all sizes from hydrogen exchange/mass spectrometry (HX-MS) data. PIGEON-FEATHER disambiguates and reconstructs all experimentally measured isotopic mass envelopes using a Bayesian Monte Carlo sampling approach. We applied PIGEON-FEATHER to reveal how E. coli and human dihydrofolate reductase orthologs (ecDHFR, hDHFR) have evolved distinct ensembles tuned to their catalytic cycles, and how two competitive inhibitors of ecDHFR arrest its ensemble in different ways. Extending the method to a large protein-DNA complex, we mapped ligand-induced ensemble reweighting in the E. coli lac repressor to understand the functional switching mechanism crucial for transcriptional regulation.
-
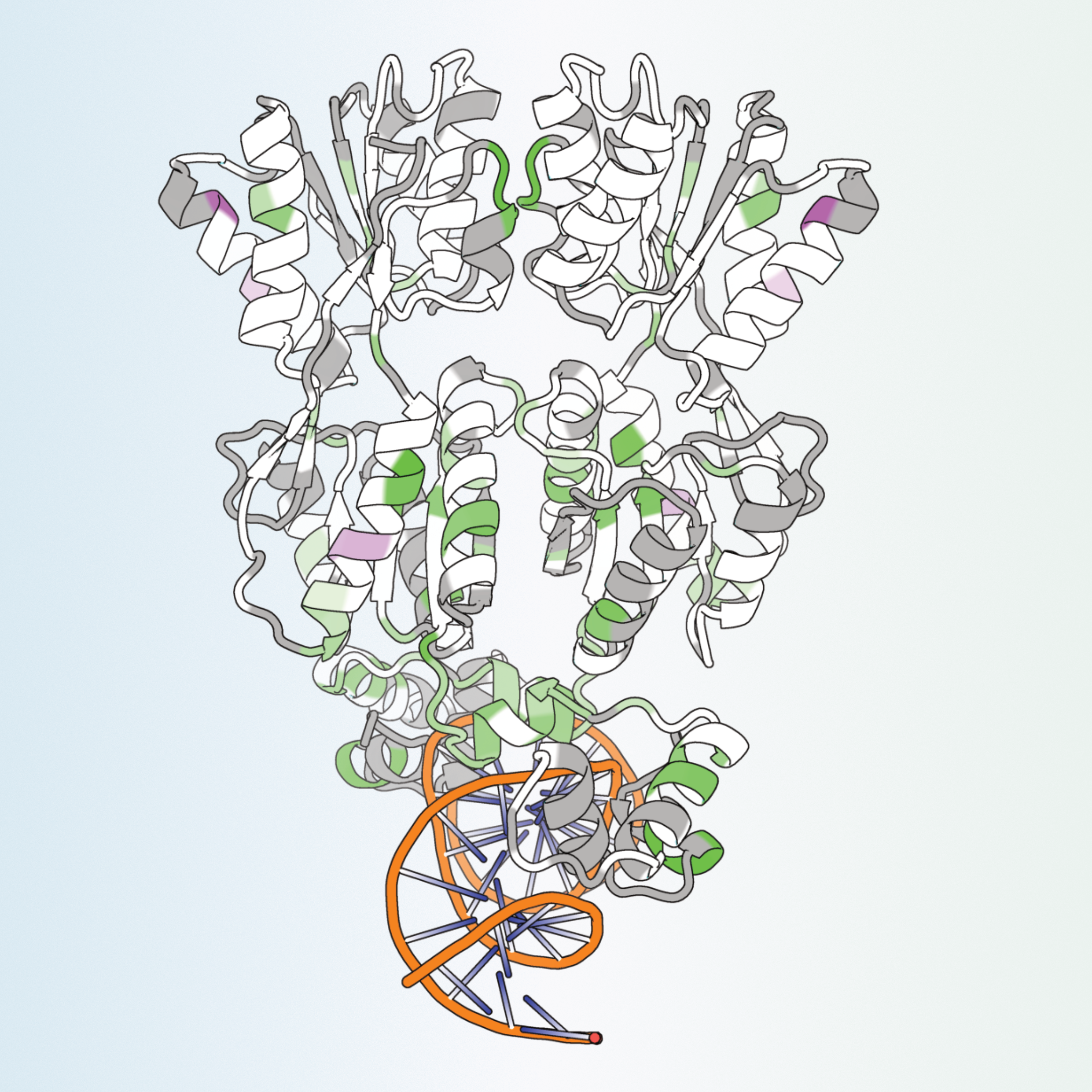 Distinct energetic blueprints diversify function of conserved protein foldsMalcolm L. Wells*, Chenlin Lu*, Daniel Sultanov*, and 3 more authorsNature Chemistry, 2026
Distinct energetic blueprints diversify function of conserved protein foldsMalcolm L. Wells*, Chenlin Lu*, Daniel Sultanov*, and 3 more authorsNature Chemistry, 2026Advances in structural biology have improved our understanding of the relationship between protein structure and function, while also confirming a widely applicable principle: protein domains with highly conserved three-dimensional folds can perform radically disparate biochemical functions. To gain insight into this structural enigma, we mapped the energetic landscapes of a family of bacterial transcription factors and their anciently diverged structural homologues, the periplasmic binding proteins. Using hydrogen exchange–mass spectrometry, bioinformatics, X-ray crystallography and molecular dynamics, we uncovered an unexpected contrast: despite binding the same sugars, the two families have evolved unique ‘energetic blueprints’ to support their distinct functional requirements. To test if differences in ensemble energies have functional consequences, we rationally redesigned the protein fold for tunable ligand-driven transcriptional responses. Strikingly, energy-driven protein engineering produced synthetic transcription factors with the theoretically anticipated ligand-induced transcriptional outputs. Thus, decoding energetic blueprints among conserved protein folds provides diverse functional adaptations, paves an alternative roadmap for protein design, and offers a distinct approach for engineering challenging drug targets.
-
 HXMS: a standardized file format for HX-MS dataKyle C. Weber*, Chenlin Lu*, R. V. Alvarez, and 2 more authorsBioinformatics, Mar 2026
HXMS: a standardized file format for HX-MS dataKyle C. Weber*, Chenlin Lu*, R. V. Alvarez, and 2 more authorsBioinformatics, Mar 2026Inspired by reliable protein structure and genomics data formats, we present HXMS, a unified, lightweight, scalable, and human-readable file format for HX-MS data. The HXMS format preserves the isotopic mass envelopes for all peptides, captures the full experimental time-course including the fully deuterated control samples, and contains all other key information. It supports multimodal distributions, post-translational modifications (PTMs), and experimental replicates. To promote compatibility with existing HX-MS workflows, we also developed PFLink, a Python package that converts exported data files from commonly used HX-MS analysis software packages to the HXMS format. PFLink and the HXMS format will enable more quantitative, higher-resolution data processing, improved data sharing and storage among HX-MS practitioners, future machine learning applications, and further developments in HX-MS analysis.
-
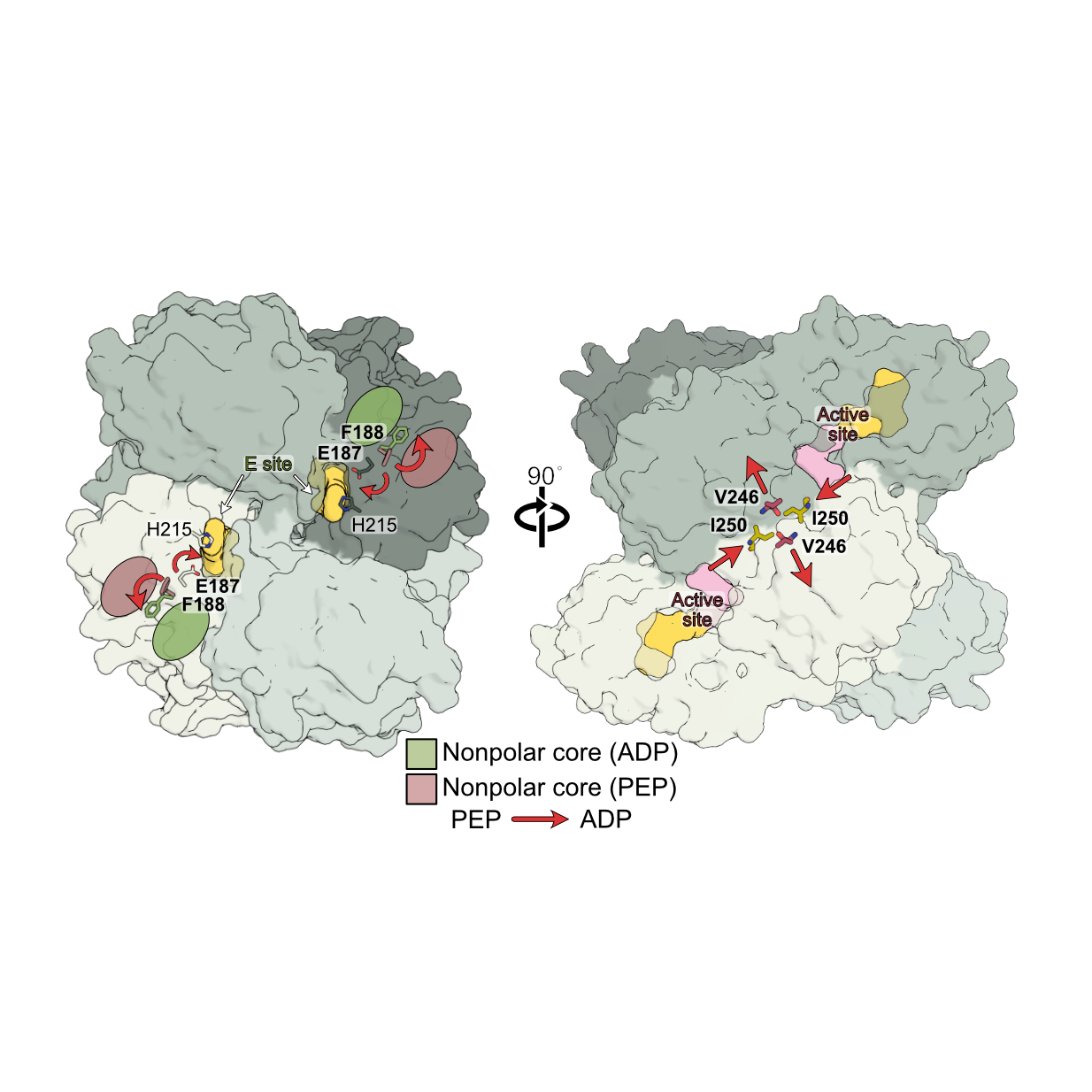 Bidirectional Allosteric Ligand Regulation in a Central Glycolytic EnzymeBelen Sundberg*, Chenlin Lu*, Malcolm L. Wells, and 3 more authorsFeb 2026Pages: 2026.02.05.704047 Section: New Results
Bidirectional Allosteric Ligand Regulation in a Central Glycolytic EnzymeBelen Sundberg*, Chenlin Lu*, Malcolm L. Wells, and 3 more authorsFeb 2026Pages: 2026.02.05.704047 Section: New ResultsAllosteric regulation enables fine-tuned control of enzyme activity in response to cellular signals, yet its molecular basis often remains unclear. Phosphofructokinase-1 (PFK), the rate-limiting enzyme of glycolysis, is a paradigmatic, well-conserved system whose reaction kinetics conform to the Monod-Wyman-Changeux model of allostery. However, X-ray crystal structures of bacterial PFK orthologs in distinct ligand-bound states do not show the consistent, concerted structural rearrangements expected for classical “relaxed” and “tense” states, revealing a decades-long disconnect between structure and function. We resolve this paradox by integrating biophysical and computational approaches to show that activator and inhibitor binding to the same allosteric pocket differentially reweight the conformational ensemble of Escherichia coli PFK. Activator binding stabilizes conformational substates that preorganize the catalytic site, whereas inhibitor binding upweights apo-like, catalytically incompetent substates. These findings establish an ensemble-based mechanism for PFK regulation and provide an energetic framework for understanding the expanded allosteric architecture of higher PFK orthologs.
2025
-
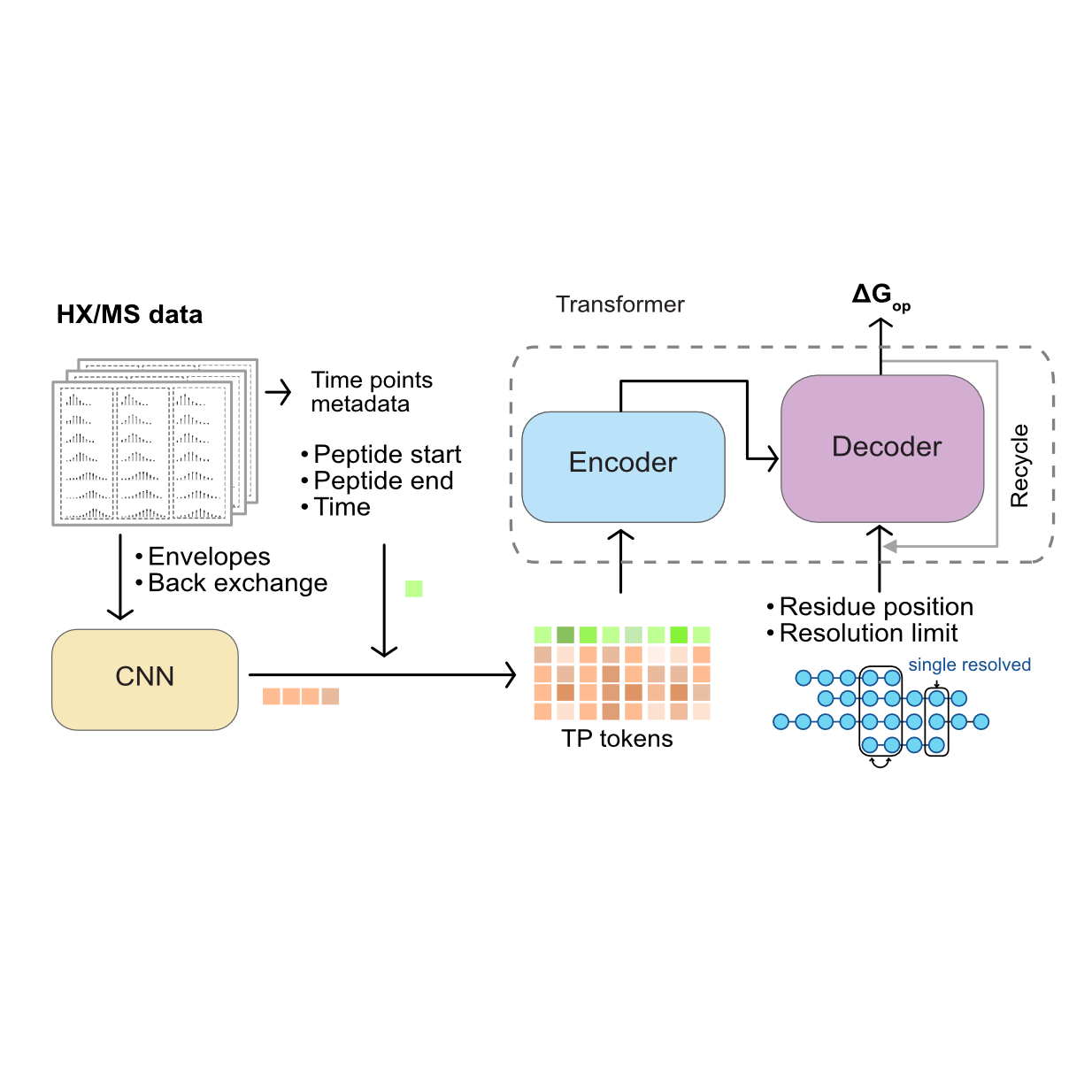 A Machine Learning Method for Calculating Highly Localized Protein StabilitiesChenlin Lu*, Kyle C. Weber*, Savannah K. McBride, and 2 more authorsOct 2025Pages: 2025.10.21.683809 Section: New Results
A Machine Learning Method for Calculating Highly Localized Protein StabilitiesChenlin Lu*, Kyle C. Weber*, Savannah K. McBride, and 2 more authorsOct 2025Pages: 2025.10.21.683809 Section: New ResultsThe residue-level free energy of opening (∆Gop) is the ultimate thermodynamic descriptor of localized protein stability, providing valuable information about the protein ensemble at physiologically relevant timescales and conditions. PFNet instantly determines ΔGop for arbitrarily large proteins and complexes from conventional peptide-level hydrogen exchange/mass spectrometry (HX-MS) datasets. It unlocks the full potential of HX-MS, democratizing the method and establishing quantitative, scalable and accessible analysis (https://github.com/glasgowlab/PFNet).
2024
-
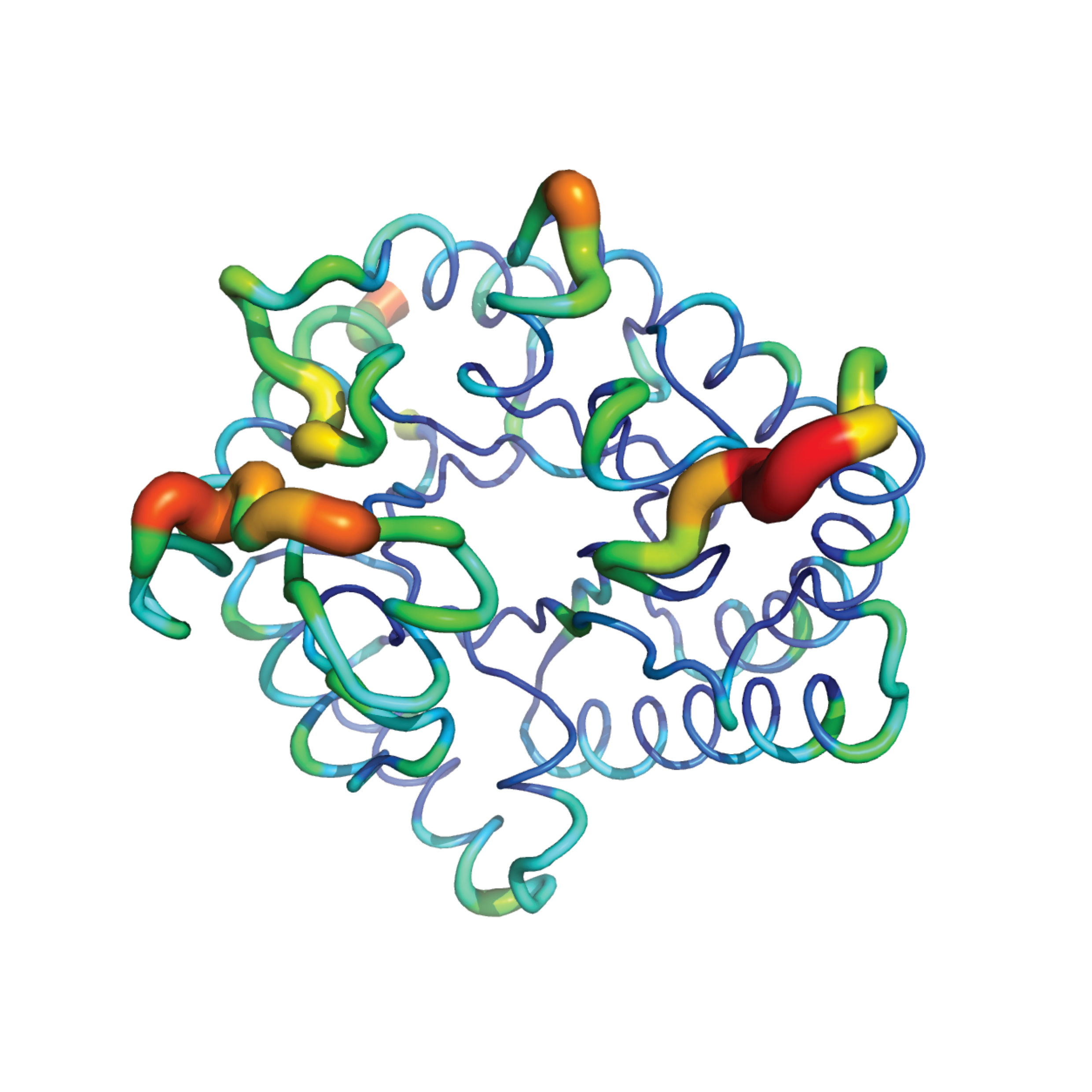 Enhancing the stability of a novel D-allulose 3-epimerase from Ruminococcus sp. CAG55 by interface interaction engineering and terminally attached a self-assembling peptideJing Wang, Chenlin Lu, Xuemei Shen, and 6 more authorsInternational Journal of Biological Macromolecules, Jun 2024
Enhancing the stability of a novel D-allulose 3-epimerase from Ruminococcus sp. CAG55 by interface interaction engineering and terminally attached a self-assembling peptideJing Wang, Chenlin Lu, Xuemei Shen, and 6 more authorsInternational Journal of Biological Macromolecules, Jun 2024D-allulose, a highly desirable sugar substitute, is primarily produced using the D-allulose 3-epimerase (DAE). However, the availability of usable DAE enzymes is limited. In this study, we discovered and engineered a novel DAE Rum55, derived from a human gut bacterium Ruminococcus sp. CAG55. The activity of Rum55 was strictly dependent on the presence of Co2+, and it exhibited an equilibrium conversion rate of 30.6 % and a half-life of 4.5 h at 50 ◦C. To enhance its performance, we engineered the interface interaction of Rum55 to stabilize its tetramer structure, and the best variant E268R was then attached with a self-assembling peptide to form active enzyme aggregates as carrier-free immobilization. The half-life of the best variant E268R-EKL16 at 50 ◦C was dramatically increased 30-fold to 135.3 h, and it maintained 90 % of its activity after 13 consecutive reaction cycles. Additionally, we identified that metal ions played a key role in stabilizing the tetramer structure of Rum55, and the dependence on metal ions for E268R-EKL16 was significantly reduced. This study provides a useful route for improving the thermostability of DAEs, opening up new possibilities for the industrial production of D-allulose.
2023
-
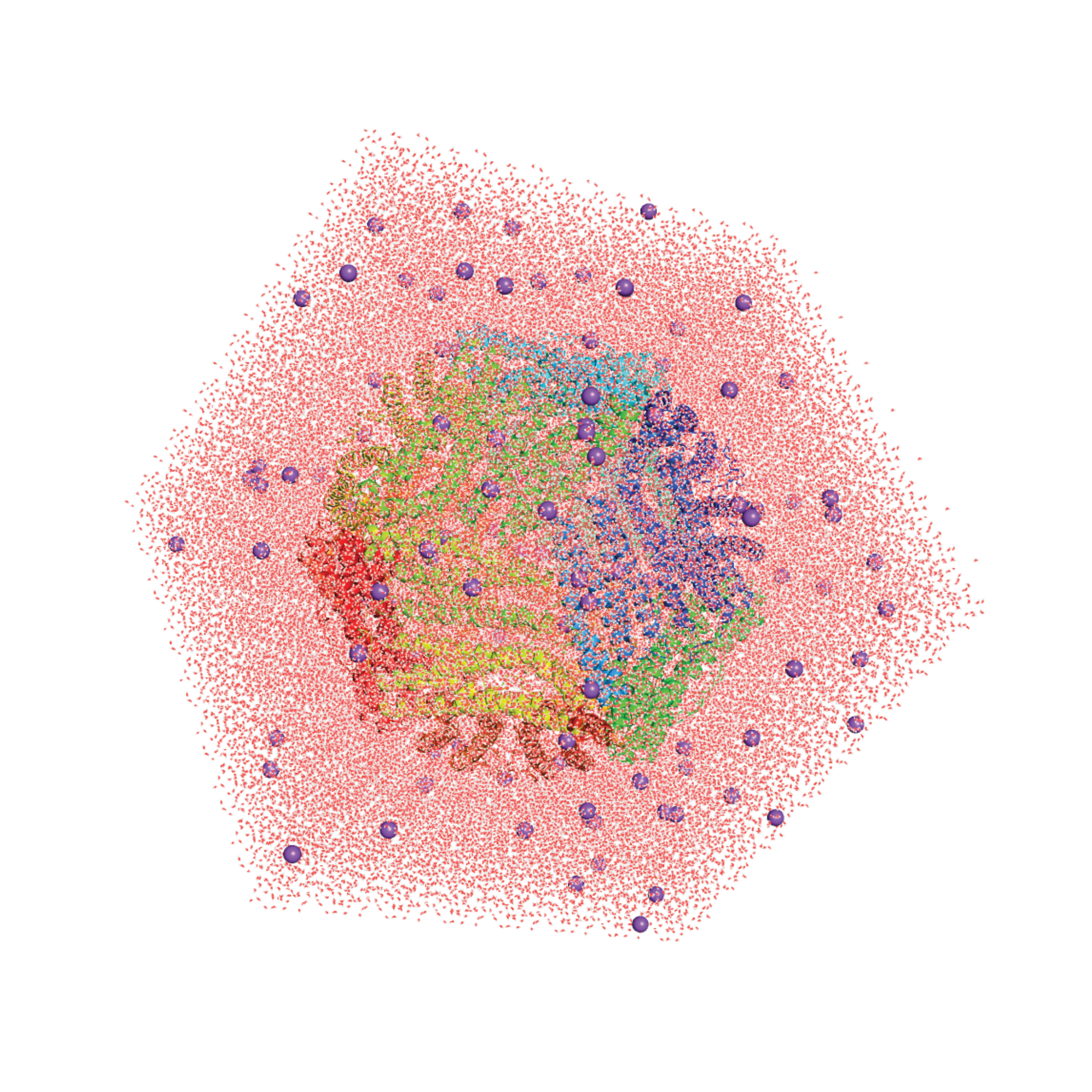 Molecular Dynamics Simulation of Protein CagesChenlin Lu, Xue Peng, and Diannan LuIn Protein Cages: Design, Structure, and Applications, Jun 2023
Molecular Dynamics Simulation of Protein CagesChenlin Lu, Xue Peng, and Diannan LuIn Protein Cages: Design, Structure, and Applications, Jun 2023Molecular dynamics (MD) simulations enable the description of the physical movement of the system over time based on classical mechanics at various scales depending on the models. Protein cages are a particular group of different-size proteins with hollow, spherical structures and are widely found in nature, which have vast applications in numerous fields. The MD simulation of cage proteins is particularly important as a powerful tool to unveil their structures and dynamics for various properties, assembly behavior, and molecular transport mechanisms. Here, we describe how to conduct MD simulations for cage proteins, especially technical details, and analyze some of the properties of interest using GROMACS/NAMD packages.
-
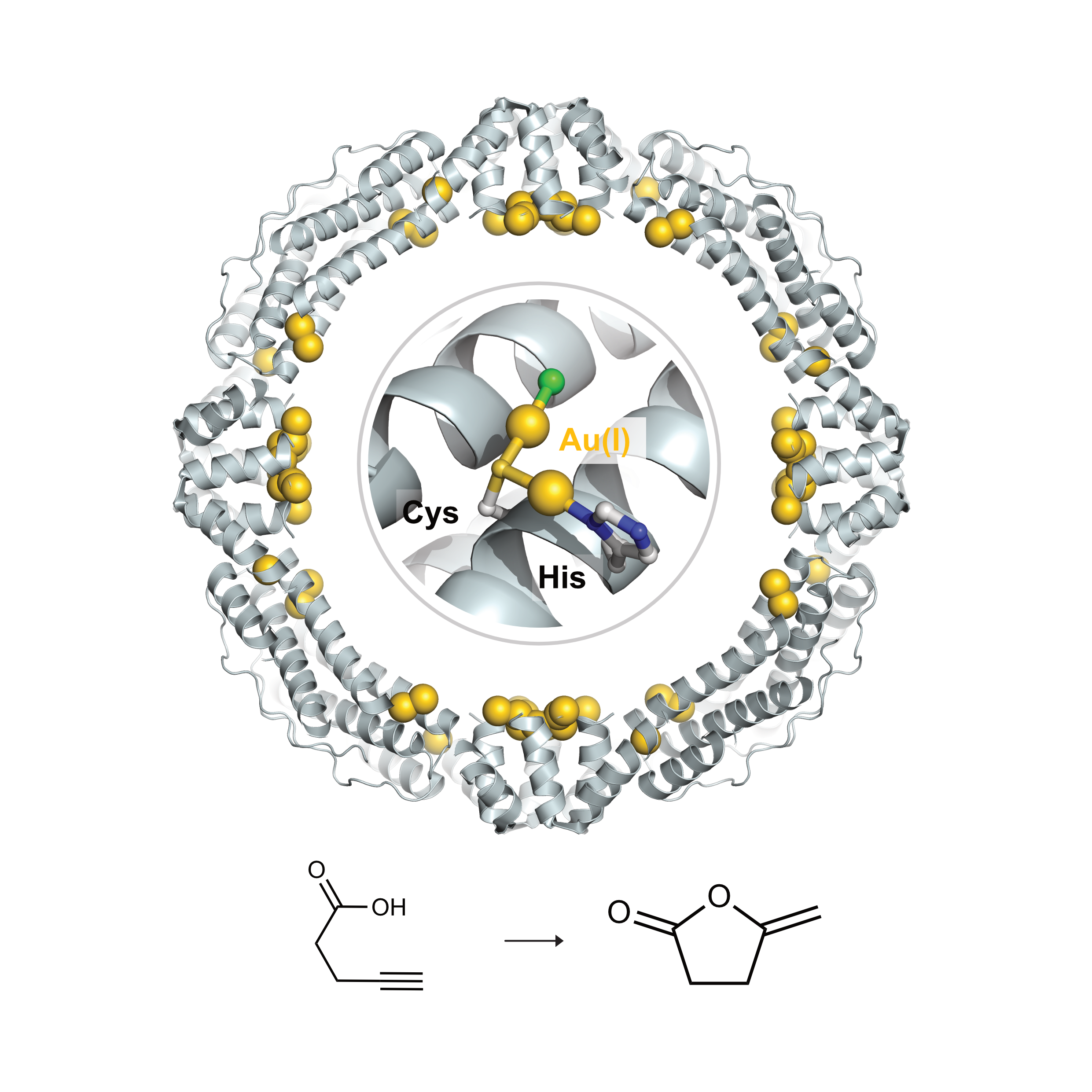 Novel Au(I)-Based Artificial Metallo-Cycloisomerase for Catalyzing the Cycloisomerization of γ-Alkynoic AcidsChenlin Lu, Xue Peng, Basudev Maity, and 5 more authorsACS Catalysis, Jul 2023
Novel Au(I)-Based Artificial Metallo-Cycloisomerase for Catalyzing the Cycloisomerization of γ-Alkynoic AcidsChenlin Lu, Xue Peng, Basudev Maity, and 5 more authorsACS Catalysis, Jul 2023Artificial metalloenzymes, which are designed rationally as hybrids of proteins and catalytically active transition-metal complexes, have become a promising approach for catalyzing unprecedented reactions for natural enzymes. In this study, we described the design and synthesis of an artificial metalloenzyme, a cycloisomerase that utilizes Au(I) incorporated into an apo-ferritin cage (Fr−Au) to efficiently catalyze the cycloisomerization of alkynoic acids, with a conversion of 83% and a turnover frequency of 20.6 × 103· h−1 in aqueous solution under mild conditions. The remarkable catalytic activity indicates that the nano-confinement of the Au(I) active site within the ferritin cage enhances its catalytic properties by stabilizing and solubilizing it. The less protected Au atom in the cysteine bridged dinuclear Au(I) active center was identified as critical for the Fr−Au cycloisomerases to catalyze this reaction. In addition, we provide insight into the catalytic mechanism through quantum chemical (QC) calculations, which reveal an energy barrier of 32.29 kJ/mol.
2022
-
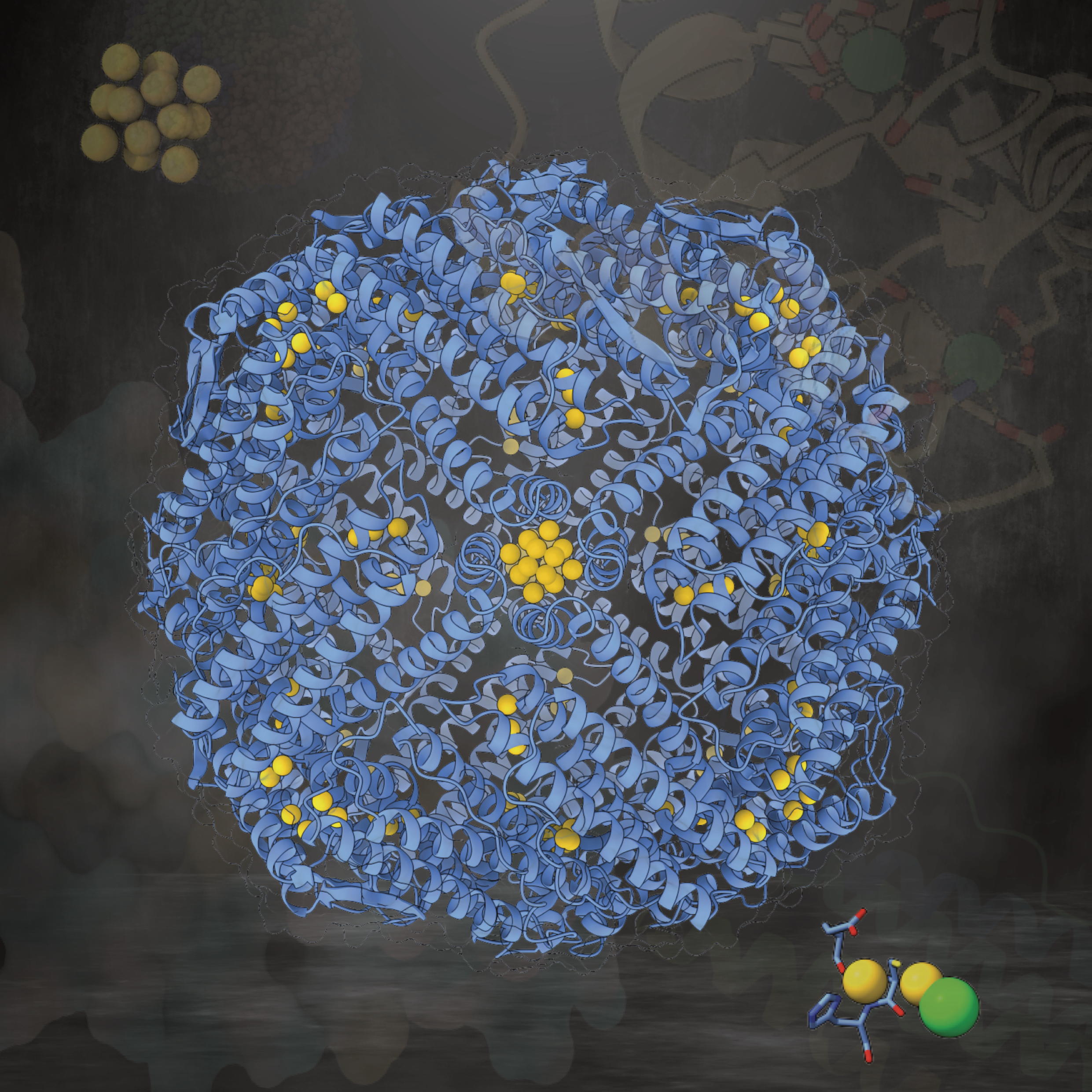 Design of a gold clustering site in an engineered apo-ferritin cageChenlin Lu, Basudev Maity, Xue Peng, and 5 more authorsCommunications Chemistry, Dec 2022
Design of a gold clustering site in an engineered apo-ferritin cageChenlin Lu, Basudev Maity, Xue Peng, and 5 more authorsCommunications Chemistry, Dec 2022Water-soluble and biocompatible protein-protected gold nanoclusters (Au NCs) hold great promise for numerous applications. However, design and precise regulation of their structure at an atomic level remain challenging. Herein, we have engineered and constructed a gold clustering site at the 4-fold symmetric axis channel of the apo-ferritin cage. Using a series of X-ray crystal structures, we evaluated the stepwise accumulation process of Au ions into the cage and the formation of a multinuclear Au cluster in our designed cavity. We also disclosed the role of key residues in the metal accumulation process. X-ray crystal structures in combination with quantum chemical (QC) calculation revealed a unique Au clustering site with up to 12 Au atoms positions in the cavity. Moreover, the structure of the gold nanocluster was precisely tuned by the dosage of the Au precursor. As the gold concentration increases, the number of Au atoms position at the clustering site increases from 8 to 12, and a structural rearrangement was observed at a higher Au concentration. Furthermore, the binding affinity order of the four Au binding sites on apo-ferritin was unveiled with a stepwise increase of Au precursor concentration.
2021
-
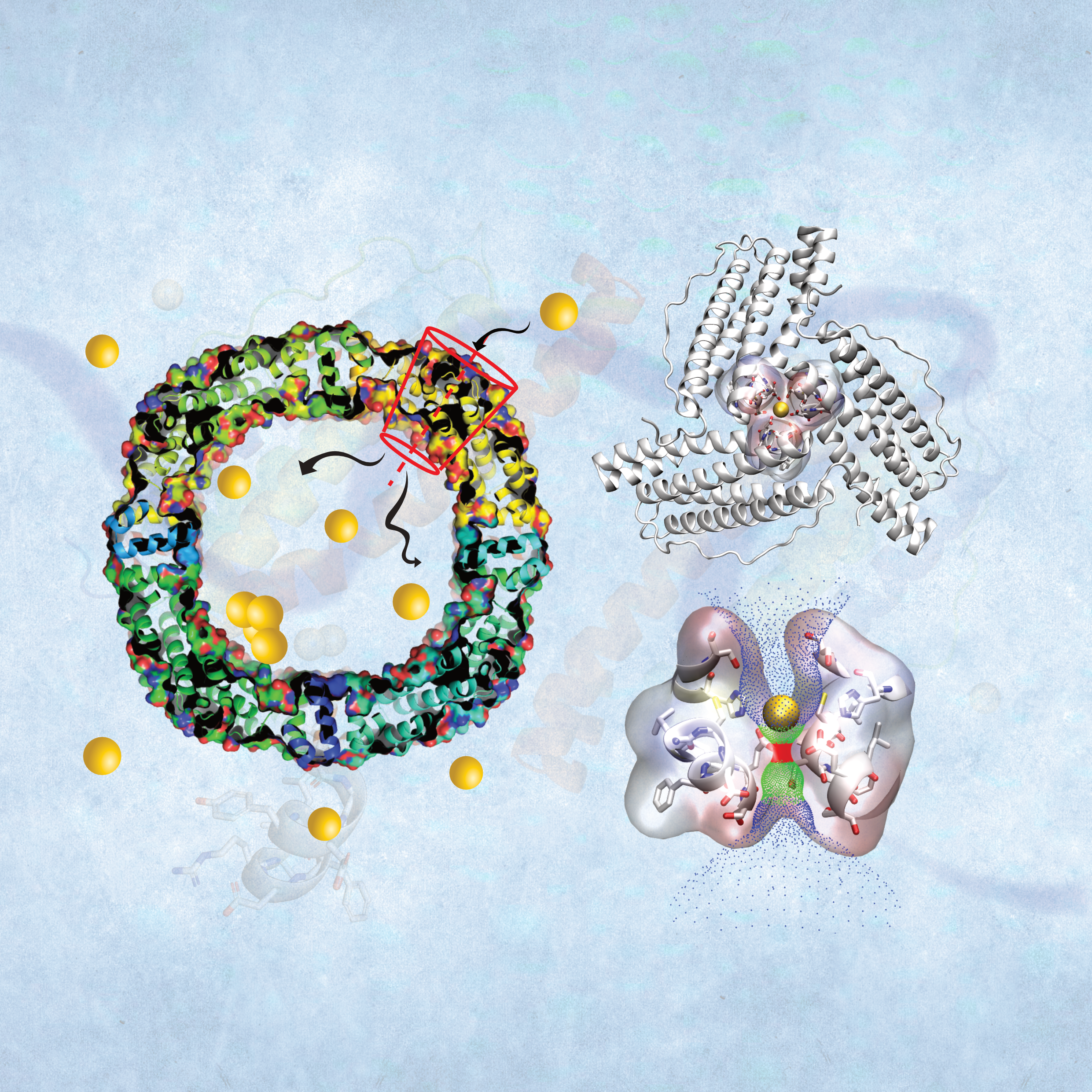 The synergistic mechanisms of apo-ferritin structural transitions and Au(III) ion transportation: molecular dynamics simulations with the Markov state modelXue Peng*, Chenlin Lu*, Zheng Liu, and 1 more authorPhysical Chemistry Chemical Physics, Dec 2021
The synergistic mechanisms of apo-ferritin structural transitions and Au(III) ion transportation: molecular dynamics simulations with the Markov state modelXue Peng*, Chenlin Lu*, Zheng Liu, and 1 more authorPhysical Chemistry Chemical Physics, Dec 2021Due to its unique structure, recent years have witnessed the use of apo-ferritin to accumulate various non-natural metal ions as a scaffold for nanomaterial synthesis. However, the transport mechanism of metal ions into the cavity of apo-ferritin is still unclear, limiting the rational design and controllable preparation of nanomaterials. Here, we conducted all-atom classical molecular dynamics (MD) simulations combined with Markov state models (MSMs) to explore the transportation behavior of Au(III) ions. We exhibited the complete transportation paths of Au(III) from solution into the apo-ferritin cage at the atomic level. We also revealed that the transportation of Au(III) ions is accompanied by coupled protein structural changes. It is shown that the 3-fold axis channel serves as the only entrance with the longest residence time of Au(III) ions. Besides, there are eight binding clusters and five 3-fold structural metastable states, which are important during Au(III) transportation. The conformational changes of His118, Asp127, and Glu130, acting as doors, were observed to highly correlate with the Au(III) ion’s position. The MSM analysis and Potential Mean Force (PMF) calculation suggest a remarkable energy barrier near Glu130, making it the rate-limiting step of the whole process. The dominant transportation pathway is from cluster 3 in the 3-fold channel to the inner cavity to cluster 5 on the inner surface, and then to cluster 6. These findings provide inspiration and theoretical guidance for the further rational design and preparation of new nanomaterials using apo-ferritin.
-
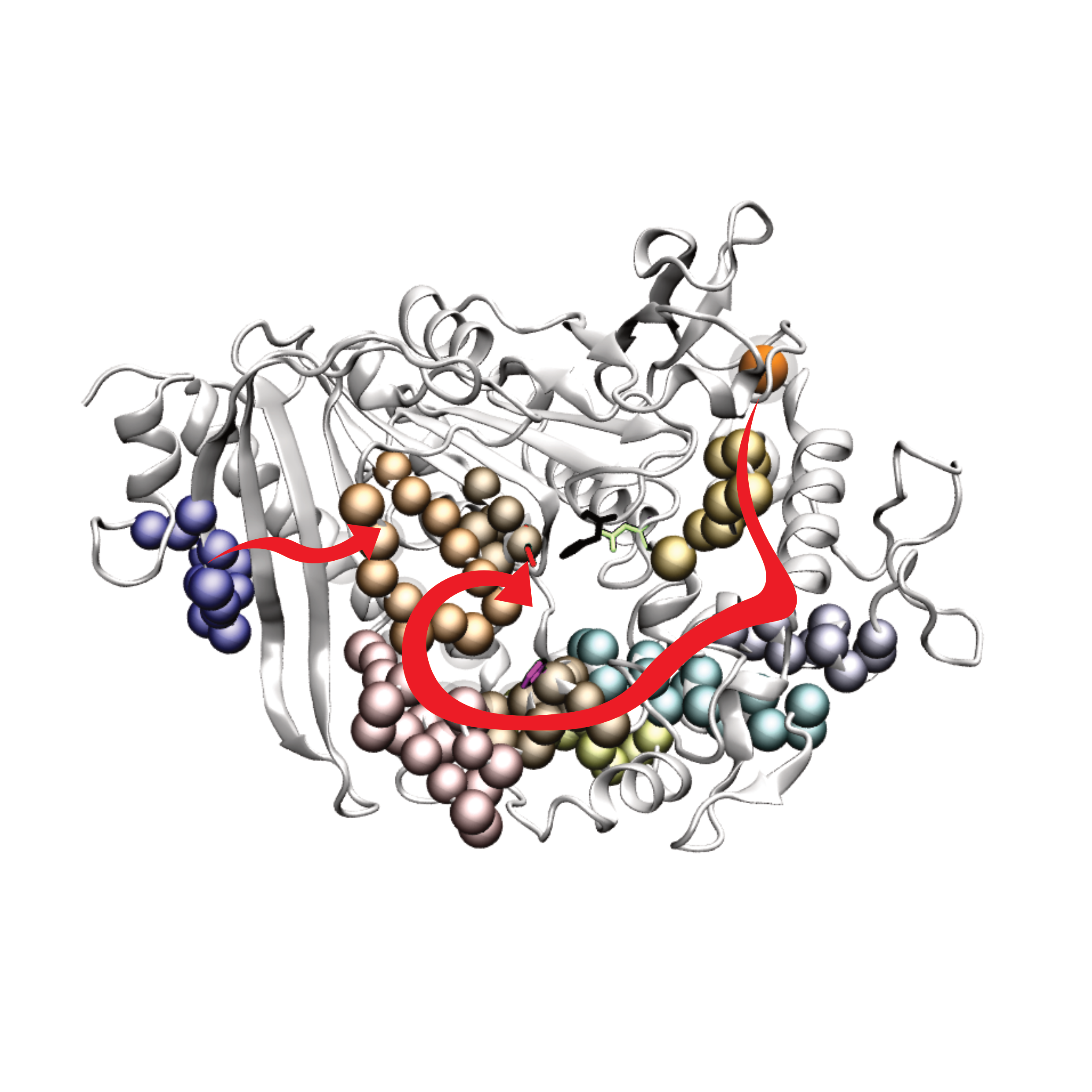 A distal regulatory strategy of enzymes: from local to global conformational dynamicsXue Peng*, Chenlin Lu*, Jian Pang, and 2 more authorsPhysical Chemistry Chemical Physics, Dec 2021
A distal regulatory strategy of enzymes: from local to global conformational dynamicsXue Peng*, Chenlin Lu*, Jian Pang, and 2 more authorsPhysical Chemistry Chemical Physics, Dec 2021Modulating the distribution of various states in protein ensembles through distal sites may be promising in the evolution of enzymes in desired directions. , Modulating the distribution of various states in protein ensembles through distal sites may be promising in the evolution of enzymes in desired directions. However, the prediction of distal mutation hotspots that stabilize the favoured states from a computational perspective remains challenging. Here, we presented a strategy based on molecular dynamics (MD) and Markov state models (MSM) to predict distal mutation sites. Extensive MD combined with MSM was applied to determine the principally distributed metastable states interconverting at a slow timescale. Then, molecular docking was used to classify these states into active states and inactive ones. Distal mutation hotspots were targeted based on comparing the conformational features between active and inactive states, where mutations destabilize the inactive states and show little influence on the active state. The proposed strategy was used to explore the highly dynamic MHETase, which shows a potential application in the biodegradation of poly(ethylene terephthalate) (PET). Seven principally populated interrelated metastable states were identified, and the atomistic picture of their conformational changes was unveiled. Several residues at distal positions were found to adopt more H-bond occupancies in inactive states than active states, making them potential mutation hotspots for stabilizing the favoured conformations. In addition, the detailed mechanism revealed the significance of calcium ions at a distance from the catalytic centre in reshaping the free energy landscape. This study deepens the understanding of the conformational dynamics of α/β hydrolases containing a lid domain and advances the study of enzymatic plastic degradation.
2020
-
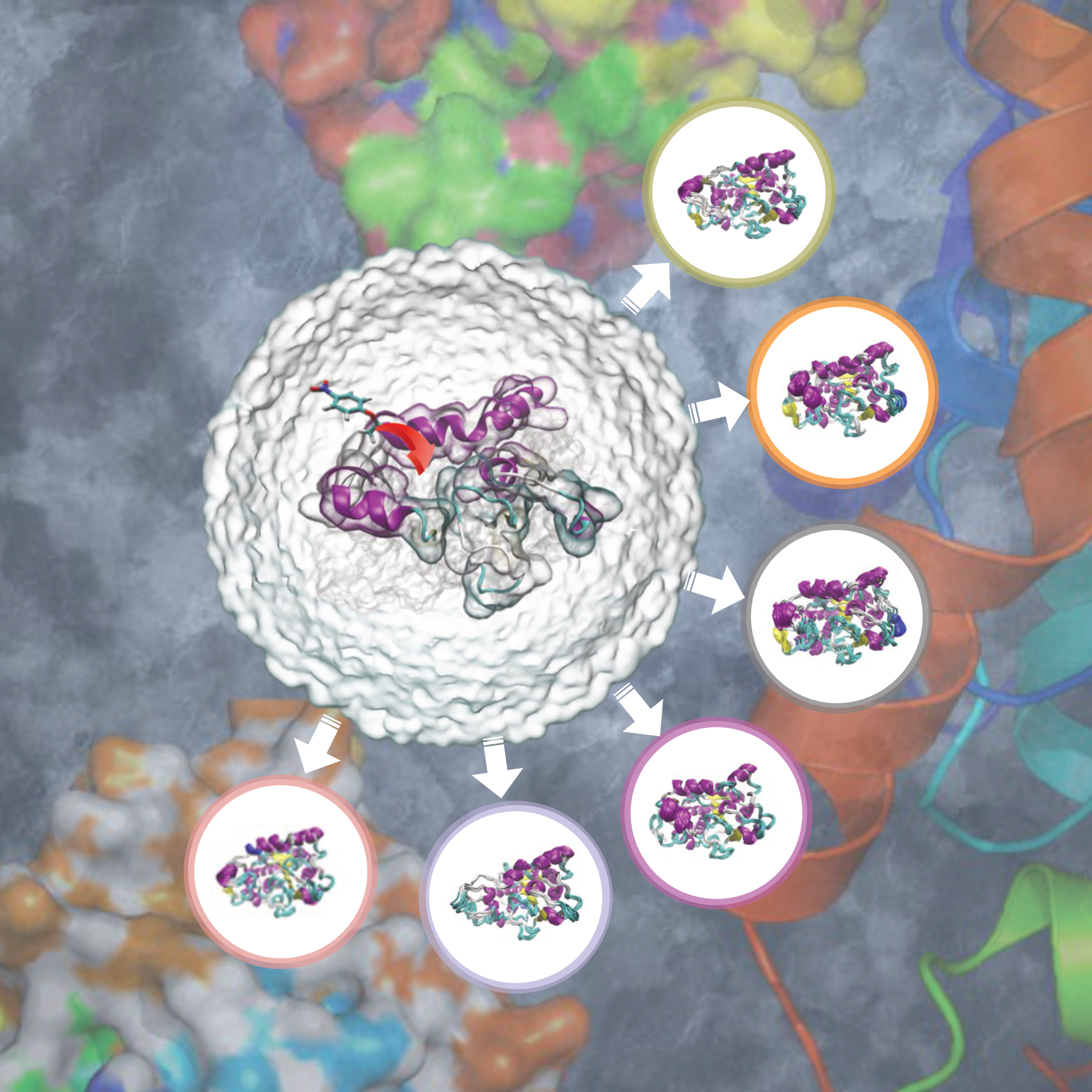 Global and Kinetic Profiles of Substrate Diffusion in \textitCandida antarctica Lipase B: Molecular Dynamics with the Markov-State ModelChenlin Lu, Xue Peng, Diannan Lu, and 1 more authorACS Omega, May 2020
Global and Kinetic Profiles of Substrate Diffusion in \textitCandida antarctica Lipase B: Molecular Dynamics with the Markov-State ModelChenlin Lu, Xue Peng, Diannan Lu, and 1 more authorACS Omega, May 2020Profiling substrate diffusion pathways with kinetic information, which accounts for the dynamic nature of enzyme−substrate interaction, can enable molecular reengineering of enzymes and process optimization of enzymatic catalysis. Candida antarctica lipase B (CALB) is extensively used for producing various chemicals because of its rich catalytic mechanisms, broad substrate spectrum, thermal stability, and tolerance to organic solvents. In this study, an all-atom molecular dynamics (MD) combined with Markov-state models (MSMs) implemented in pyEMMA was proposed to simulate diffusion pathways of 4-nitrophenyl ester (4NPE), a commonly used substrate, from the surface into the active site of CALB. Six important metastable conformations of CALB were identified in the diffusion process, including a closed state. An induced-fit mechanism incorporating multiple pathways with molecular information was proposed, which might find unprecedented applications for the rational design of lipase for green catalysis.